熱線:021-66110810,66110819
手機:13564362870
熱線:021-66110810,66110819
手機:13564362870
Mitochondrial dysfunction underlies impaired neurovascular coupling following traumatic brain injury
線粒體功能障礙是創傷性腦損傷后神經血管耦合受損的基礎
期刊:Neurobiology of Disease
來源:ScienceDirect
摘要核心內容
本研究探討創傷性腦損傷(TBI)后繼發性損傷的機制,重點關注線粒體功能障礙與神經血管耦合(NVC)受損的關系。通過閉合性顱腦損傷大鼠模型,研究發現:
TBI后皮層擴散性抑制(CSD)和癲癇發作會加劇線粒體損傷(如線粒體嵴破壞、活性氧(ROS)增加)。
線粒體功能障礙導致血管調節異常,表現為CSD后血管收縮加劇和局部腦血流(CBF)減少。
這些變化與神經行為學衰退相關,表明代謝應激是TBI后繼發性損傷的關鍵驅動因素。
研究目的
探究TBI后線粒體損傷如何影響神經血管單元(NVU)功能,特別是在代謝挑戰(如CSD和癲癇)下,揭示線粒體功能障礙與神經血管耦合異常之間的因果關系。
研究思路
模型建立:采用大鼠閉合性顱腦損傷模型(450g重物從1.02m高度下落)模擬中度TBI。
功能評估:通過神經行為學評分(NSS)、腦氧飽和度(spO?)和電皮質圖(ECoG)監測急性損傷反應。
干預與測量:
電刺激誘導CSD或局部應用4-氨基吡啶誘導癲癇。
同步監測局部CBF(激光多普勒血流儀)、皮層氧分壓(Unisense微電極)、線粒體ROS(MitoSOX)和鈣離子(Rhod2-AM)。
形態學分析:透射電鏡觀察血管超微結構和線粒體形態。
機制驗證:對比TBI組與對照組的代謝、血管及行為學差異。
測量數據及其意義
(1)生理與行為數據
spO?(圖1B):TBI后1分鐘腦氧飽和度顯著下降(p=0.005),反映急性缺氧應激。
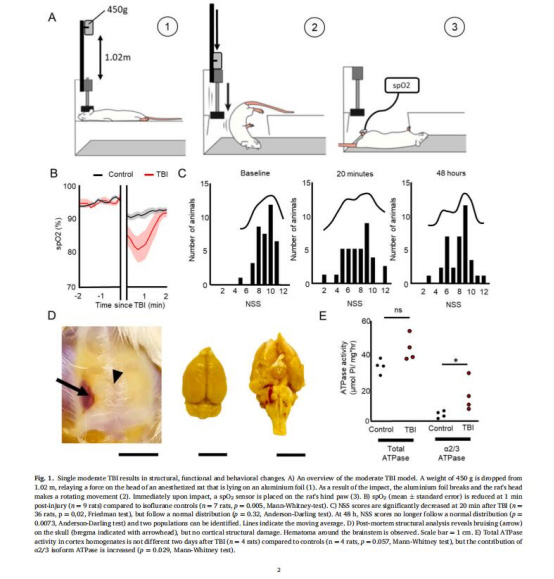
NSS評分(圖1C, 圖2D-E):
TBI后20分鐘評分降低(行為衰退),48小時后呈雙峰分布(p=0.0073),提示個體差異。
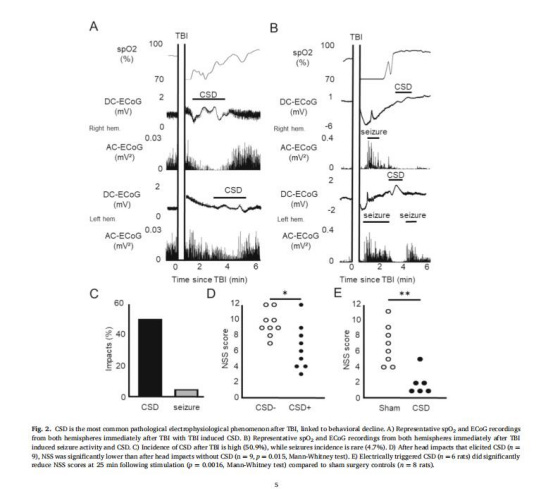
CSD直接導致NSS下降(p=0.015),關聯CSD與神經功能損傷。
ATP酶活性(圖1E):TBI后皮層α2/3亞型ATP酶活性升高(p=0.029),表明能量代謝代償性調整。
(2)電生理與血流動力學數據
CSD發生率(圖2C):TBI后CSD發生率達50.9%,癲癇僅4.7%,凸顯CSD的核心病理作用。
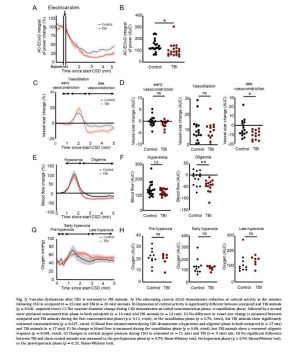
CBF與血管直徑(圖3C-F):
TBI組CSD后血管收縮更顯著(p=0.017),CBF降幅更大(p=0.004),提示血管調節功能受損。
皮層氧分壓(圖3G-H):
使用丹麥Unisense微電極測量顯示,CSD期間氧分壓先降后升,但TBI組峰值延遲(p=0.035),反映線粒體氧利用障礙。
(3)線粒體功能數據
MitoSOX熒光(圖4C-E):
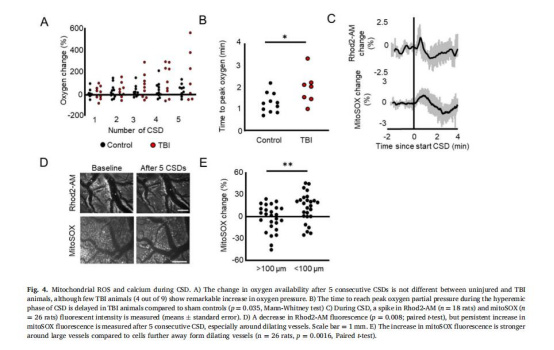
CSD后線粒體ROS持續升高,近血管區增幅更顯著(p=0.0016),表明血管周圍氧化應激加劇。
電鏡超微結構(圖5-6):
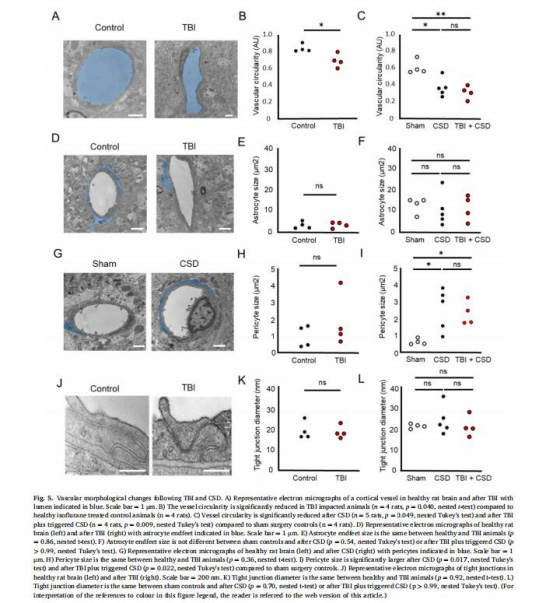
TBI后線粒體嵴損傷見于星形膠質細胞和周細胞(圖6C);CSD誘導線粒體腫脹(神經元和星形膠質細胞,圖6D-E)。


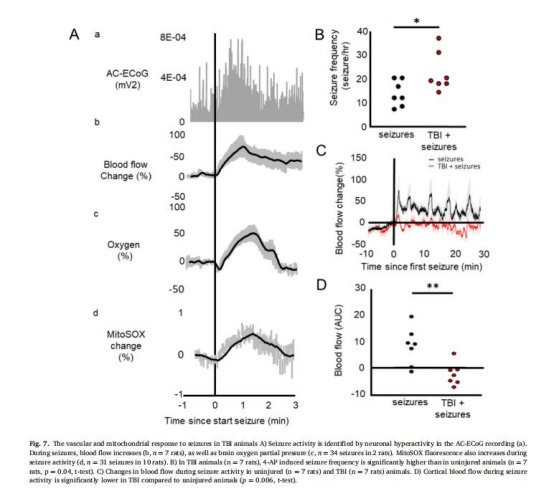
(4)癲癇模型數據(圖7)
4-AP誘導癲癇:TBI組癲癇頻率更高(p=0.04),CBF增幅更低(p=0.006),進一步證實TBI削弱代謝需求下的血管應答能力。
研究結論
CSD是TBI后繼發性損傷的關鍵媒介:通過誘發線粒體ROS暴發和鈣超載,破壞NVU功能。
線粒體損傷驅動血管功能障礙:星形膠質細胞和周細胞的線粒體嵴破壞導致CSD后病理性血管收縮及CBF減少。
代謝-血管解耦聯加重神經損傷:TBI后腦組織無法在癲癇或CSD期間有效增加氧輸送,加劇神經元損傷和行衰退。
干預靶點:靶向線粒體保護或ROS清除可能改善TBI后神經血管功能。
Unisense微電極數據的詳細解讀
測量結果(圖3G-H, 圖4A-B)
雙相氧分壓變化:CSD期間氧分壓先短暫下降(組織耗氧增加),隨后升高(血流代償性灌注)。
TBI特異性改變:
峰值延遲:TBI組氧分壓達峰時間較對照組顯著滯后(p=0.035),表明線粒體氧化磷酸化效率降低。
異常高氧:4/9 TBI動物氧分壓持續上升(線性回歸 p=0.006),提示線粒體功能障礙致氧利用率下降。
研究意義
揭示代謝-血流匹配機制:Unisense微電極首次在體捕捉到TBI后CSD期間的動態氧代謝變化,證明線粒體功能障礙直接導致神經血管解耦聯。
提供病理標志物:氧分壓變化模式可作為TBI嚴重度及治療響應的生物標志物。
指導靶向治療:數據支持開發改善線粒體氧利用的藥物(如抗氧化劑),以恢復神經血管耦合。
總結:本研究通過多模態方法證實TBI誘導線粒體損傷,進而破壞神經血管耦合,導致繼發性腦損傷。丹麥Unisense微電極的數據為核心機制(代謝-血流失匹配)提供了直接實驗證據,為開發針對線粒體的神經保護策略奠定基礎。
<strike id="yiuck"></strike>
<strike id="yiuck"></strike>