熱線:021-66110810,66110819
手機(jī):13564362870
熱線:021-66110810,66110819
手機(jī):13564362870
Mitochondrial rewiring drives metabolic adaptation to NAD(H) shortage in triple negative breast cancer cells
線粒體重新布線驅(qū)動(dòng)三陰性乳腺癌細(xì)胞對 NADH短缺的代謝適應(yīng)
期刊:Neoplasia
來源:ScienceDirect (DOI: https://doi.org/10.1016/j.neo.2023.100903)
摘要核心內(nèi)容
本研究探討三陰性乳腺癌(TNBC)細(xì)胞對NAMPT抑制劑FK866耐藥的代謝適應(yīng)機(jī)制。通過建立FK866耐藥細(xì)胞模型(MDA-MB-231 RES),研究發(fā)現(xiàn):
耐藥機(jī)制獨(dú)立于經(jīng)典藥泵外排或補(bǔ)償性NAD?合成通路(如NMRK1),而依賴線粒體代謝重組。
線粒體功能增強(qiáng):耐藥細(xì)胞線粒體備用呼吸能力(SRC)、線粒體質(zhì)量(mtDNA/nDNA比例增加)及生物合成(PGC-1α/TFAM上調(diào))顯著提升。
代謝底物依賴性改變:耐藥細(xì)胞更依賴丙酮酸和琥珀酸氧化供能,且線粒體丙酮酸載體MPC2的抑制可誘導(dǎo)敏感細(xì)胞產(chǎn)生類耐藥表型。
關(guān)鍵結(jié)論:持續(xù)NAD(H)短缺驅(qū)動(dòng)線粒體重編程,通過增強(qiáng)氧化磷酸化(OXPHOS)和底物靈活性(丙酮酸→琥珀酸轉(zhuǎn)換)維持能量穩(wěn)態(tài),導(dǎo)致FK866耐藥。
研究目的
探究TNBC細(xì)胞對NAMPT抑制劑FK866耐藥的分子機(jī)制,重點(diǎn)關(guān)注線粒體代謝適應(yīng)在抵抗NAD(H)短缺中的作用,為克服耐藥提供新靶點(diǎn)。
研究思路
模型建立:
通過長期遞增FK866暴露,構(gòu)建MDA-MB-231耐藥細(xì)胞系(RES)。
耐藥機(jī)制初篩:
排除NAMPT突變、藥泵外排(ABCB1/ABCC1)及補(bǔ)償性NAD?合成通路(NMRK1沉默驗(yàn)證)。
線粒體功能聚焦:
Seahorse分析糖酵解率(GlycoPER)和線粒體呼吸功能(OCR)。
評(píng)估線粒體質(zhì)量(mtDNA/nDNA比例、MitoTracker染色)、生物合成基因(PGC-1α/TFAM)及底物依賴性(Mitoplate S-1)。
關(guān)鍵靶點(diǎn)驗(yàn)證:
藥理學(xué)(UK5099/羅格列酮抑制MPC)和遺傳學(xué)(siRNA沉默MPC1/MPC2)干預(yù),結(jié)合FK866處理。
呼吸鏈復(fù)合物活性及ATP合成酶功能檢測。
機(jī)制整合:
解析丙酮酸→琥珀酸代謝轉(zhuǎn)換在維持膜電位(Δψm)和能量供應(yīng)中的作用。
測量數(shù)據(jù)及其意義
(1)耐藥表型與機(jī)制排除(圖1)
數(shù)據(jù):RES細(xì)胞對FK866完全耐藥(EC??不可計(jì)算),但對化療藥物(5FU、順鉑)敏感;Verapamil/CsA處理不逆轉(zhuǎn)耐藥;ABCB1/ABCC1表達(dá)無差異。
意義:耐藥非經(jīng)典藥泵介導(dǎo),提示代謝適應(yīng)主導(dǎo)。
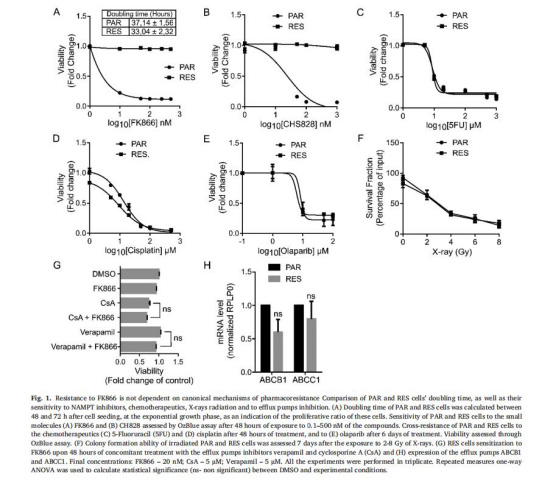 來源:圖1A-G。
來源:圖1A-G。
(2)NAD?合成通路無補(bǔ)償作用(圖2)
數(shù)據(jù):RES細(xì)胞NAMPT/NAPRT下調(diào)、NMRK1上調(diào),但NMRK1沉默不增敏FK866;NAD(H)/ATP水平不變。
意義:補(bǔ)償性NAD?合成非關(guān)鍵耐藥機(jī)制。

來源:圖2B-H。
(3)線粒體功能增強(qiáng)(圖3)
數(shù)據(jù):
RES細(xì)胞備用呼吸容量(SRC) 顯著升高(Seahorse OCR)。
線粒體質(zhì)量增加:mtDNA/nDNA比例↑、TOMM20蛋白↑、PGC-1α/TFAM轉(zhuǎn)錄上調(diào)、MitoTracker熒光強(qiáng)度↑2倍。
意義:線粒體生物合成與功能擴(kuò)容是核心適應(yīng)策略。
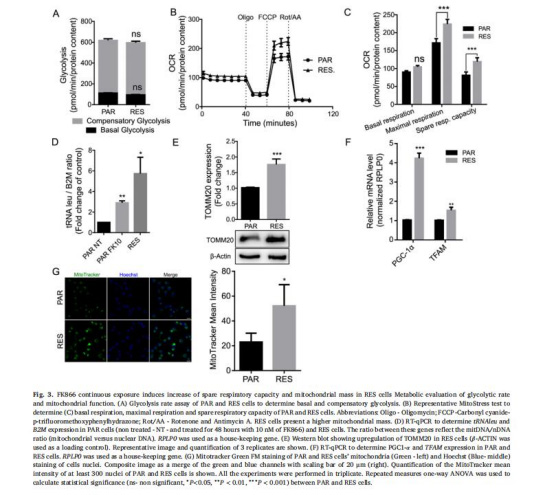
來源:圖3A-G。
(4)代謝底物依賴性轉(zhuǎn)變(圖4)
數(shù)據(jù):Mitoplate分析顯示RES細(xì)胞對丙酮酸+低劑量蘋果酸氧化速率↑;UK5099(MPC抑制劑)降低RES細(xì)胞最大呼吸。
意義:耐藥細(xì)胞依賴丙酮酸作為碳源,MPC介導(dǎo)的丙酮酸輸入至關(guān)重要。
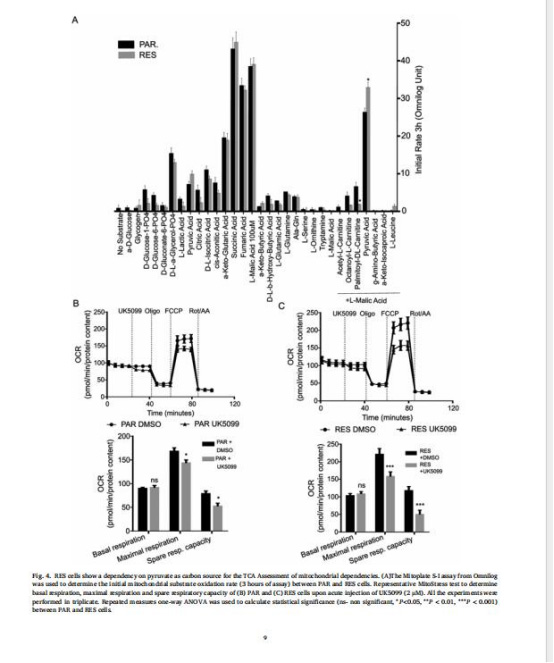
來源:圖4A-C。
(5)MPC2抑制誘導(dǎo)類耐藥表型(圖5)
數(shù)據(jù):
UK5099/羅格列酮聯(lián)合FK866提升敏感細(xì)胞存活率。
MPC2沉默(非MPC1)部分挽救FK866導(dǎo)致的細(xì)胞死亡和ATP耗竭。
意義:MPC2功能抑制模擬耐藥表型,驗(yàn)證其介導(dǎo)代謝適應(yīng)的必要性。
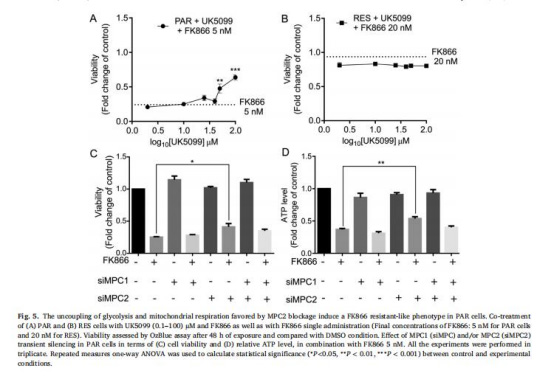
來源:圖5A-D。
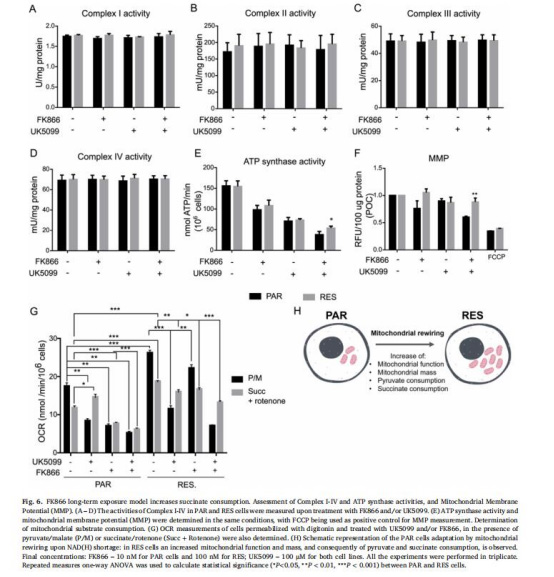
(6)琥珀酸依賴性與能量維持(圖6)
數(shù)據(jù):
呼吸鏈復(fù)合物I-IV活性無差異,但ATP合成酶活性在FK866+UK5099處理下RES細(xì)胞更高。
丹麥Unisense微電極數(shù)據(jù):RES細(xì)胞在丙酮酸剝奪(UK5099)下,琥珀酸氧化耗氧率(OCR) 顯著高于敏感細(xì)胞(圖6G)。
意義:耐藥細(xì)胞通過琥珀酸氧化維持膜電位(Δψm)和ATP合成,提供代謝靈活性。
來源:圖6A-G。
研究結(jié)論
線粒體代謝重組是耐藥核心:FK866持續(xù)暴露誘導(dǎo)線粒體生物合成(質(zhì)量↑)和功能擴(kuò)容(SRC↑),增強(qiáng)OXPHOS能力。
底物靈活性驅(qū)動(dòng)適應(yīng):耐藥細(xì)胞從丙酮酸依賴轉(zhuǎn)向琥珀酸高效利用(Unisense數(shù)據(jù)證實(shí)),維持能量穩(wěn)態(tài)。
MPC2是關(guān)鍵調(diào)控點(diǎn):抑制MPC2(非MPC1)誘導(dǎo)敏感細(xì)胞產(chǎn)生類耐藥表型,靶向MPC2或可逆轉(zhuǎn)化療耐藥。
丹麥Unisense微電極數(shù)據(jù)的詳細(xì)解讀
測量方法與結(jié)果(圖6G)
技術(shù)原理:Unisense微電極通過實(shí)時(shí)耗氧率(OCR) 檢測,評(píng)估線粒體底物氧化效率。細(xì)胞經(jīng)皂苷透化后,分別提供:
丙酮酸+蘋果酸(激活復(fù)合物I通路)。
琥珀酸+魚藤酮(激活復(fù)合物II通路,抑制復(fù)合物I)。
關(guān)鍵發(fā)現(xiàn):
耐藥細(xì)胞琥珀酸OCR更高:RES細(xì)胞在琥珀酸底物下OCR顯著高于PAR細(xì)胞(*p<0.01),表明其增強(qiáng)琥珀酸氧化能力。
代謝靈活性:UK5099抑制丙酮酸輸入后,RES細(xì)胞仍通過琥珀酸維持高OCR;FK866處理不影響其琥珀酸利用效率。
敏感細(xì)胞的補(bǔ)償失敗:PAR細(xì)胞在丙酮酸剝奪后琥珀酸OCR無代償性提升,且FK866進(jìn)一步抑制呼吸功能。
研究意義
揭示代謝可塑性:Unisense數(shù)據(jù)首次直接證明耐藥細(xì)胞通過琥珀酸氧化繞過丙酮酸短缺,維持線粒體呼吸,解釋其能量供應(yīng)韌性。
提供耐藥新靶點(diǎn):琥珀酸代謝途徑(如復(fù)合物II)或可成為克服NAMPT抑制劑耐藥的新干預(yù)方向。
技術(shù)優(yōu)勢:Unisense微電極提供實(shí)時(shí)動(dòng)態(tài)呼吸功能數(shù)據(jù),彌補(bǔ)Seahorse靜態(tài)分析的不足,精準(zhǔn)捕捉底物轉(zhuǎn)換的代謝適應(yīng)性。
總結(jié):本研究闡明TNBC細(xì)胞通過線粒體重編程(生物合成增強(qiáng)、底物靈活性提升)抵抗NAD(H)短缺導(dǎo)致的FK866毒性。丹麥Unisense微電極數(shù)據(jù)為核心機(jī)制(琥珀酸依賴)提供直接證據(jù),為靶向線粒體代謝逆轉(zhuǎn)耐藥提供理論依據(jù)。
<strike id="yiuck"></strike>
<strike id="yiuck"></strike>